hdWGCNA 分析
前言
IMPORTANT
hdWGCNA (hierarchical dictionary Weighted Gene Co-expression Network Analysis) 是单细胞转录组学中用于构建基因共表达网络和识别功能模块的重要工具。它通过整合细胞类型特异性的基因表达模式,帮助研究者理解细胞异质性背后的基因调控机制,识别细胞类型或状态特异性的功能基因模块。
在单细胞研究中,我们不仅关注单个基因的表达差异,更希望理解基因之间的协同调控关系。hdWGCNA 通过构建基因共表达网络,识别功能相关的基因模块,为解析细胞功能和发育提供重要线索。
hdWGCNA 的核心功能
- 基因共表达网络构建:基于基因表达相关性构建细胞类型特异性的基因共表达网络
- 功能模块识别:识别在特定细胞类型中协同表达的基因模块
- 模块特征分析:计算模块特征基因 (Module Eigengenes) 并评估其生物学意义
- 模块特异性分析:识别在特定细胞类型中特异性表达的基因模块
本篇文档旨在为单细胞研究者提供一份详尽的 hdWGCNA 技术指南,内容涵盖其基本原理、在 SeekSoul Online 云平台上的操作方法、结果解读、实战案例及常见问题,帮助您快速掌握并应用该工具。
hdWGCNA 理论基础
核心原理
hdWGCNA 的核心思想是:通过识别基因间的共表达模式,构建细胞类型特异性的基因共表达网络,并识别功能相关的基因模块。这一过程可以概括为以下几个主要步骤:
- 基因选择:根据基因表达特征选择适合进行共表达分析的基因集合
- 网络构建:基于基因表达相关性构建加权基因共表达网络
- 模块识别:通过层次聚类等方法识别网络中的基因模块
- 模块特征分析:计算每个模块的特征基因 (Module Eigengenes)
- 生物学意义评估:通过富集分析等方法评估模块的生物学功能
关键算法详解
加权基因共表达网络构建
- 原理:基于基因表达相关性计算基因间的连接强度,构建加权无向图
- 方法:使用软阈值法将基因表达相关性转换为连接强度,公式为
- 优势:相比无权网络,加权网络能更好地保留基因间的相关性信息
模块识别
- 原理:通过动态树切割算法识别基因模块
- 方法:
- 计算模块邻接矩阵
- 进行层次聚类
- 使用动态树切割算法识别聚类分支
- 为每个模块分配颜色标识
模块特征基因 (Module Eigengenes)
- 定义:模块中所有基因表达的第一主成分
- 意义:代表整个模块的表达特征
- 应用:用于模块间相关性分析和下游生物学分析
模块生物学意义评估
功能富集分析
- 方法:对每个模块中的基因进行 GO/KEGG 等功能富集分析
- 应用:推断模块的潜在生物学功能
模块与细胞类型关联分析
- 方法:计算模块特征基因与细胞类型标记基因的重叠
- 应用:识别细胞类型特异性的功能模块
云平台操作指南
在云平台上,hdWGCNA 分析流程被设计得直观易用。您无需编写代码,只需通过参数配置界面即可完成分析。
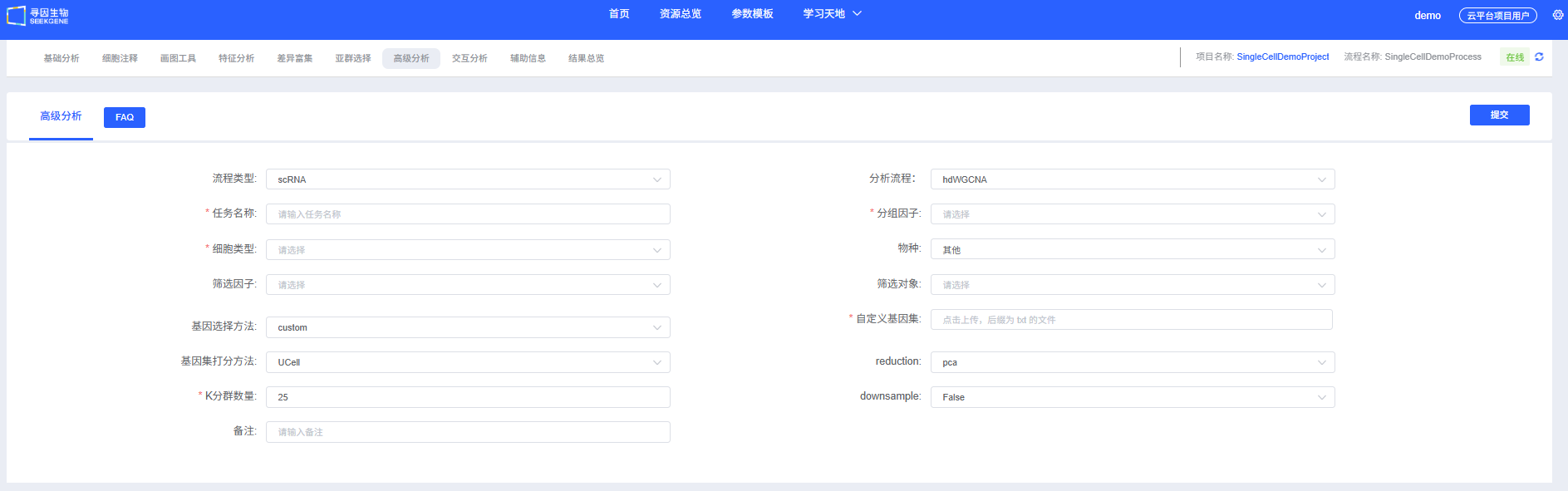
分析前的准备
IMPORTANT
hdWGCNA 分析的成功与否,很大程度上取决于输入数据的质量和生物学问题的合理性。在开始分析前,请务必确认:
- 数据已完成预处理:您的单细胞数据已经过标准的质控、降维、聚类和细胞类型注释。
- 选择了合适的细胞亚群:hdWGCNA 分析应在具有生物学意义的细胞亚群中进行,如已注释的细胞类型或功能相关的细胞簇。
- 确保数据规模适中:对于超过数万个细胞的数据集,建议开启 Downsample 进行分析以避免内存不足。
参数详解
下表详细列出了云平台 hdWGCNA 分析模块的主要参数及其说明。
| 界面参数 | 说明 |
|---|---|
| 任务名称 | 本次分析的任务名称,需以英文字母开头,可包含英文字母、数字、下划线和中文。 |
| 分组因子 | meta 的列名,和其他流程分组因子一致,例如:Cellannotation,必填。 |
| 细胞类型 | 基于分组因子的 meta 列所对应的对象,例如:T、B...,必填。 |
| 物种 | human|其他。 |
| 筛选因子 | meta 的列名,和其他流程分组因子一致,非必填,与分组因子互斥。 |
| 筛选对象 | 基于筛选因子的 meta 列所对应的对象,可多选,非必填。 |
| 基因选择方法 | 共表达网络分析用基因的选择方法:variable|fraction|custom - variable:使用存储在 Seurat 对象中的高变基因 - fraction:使用在某组中以一定比例的细胞表达的基因 - custom:自定义的基因集,选择后 gene_list 必须指定 |
| 基因选择比例 | 当基因选择方法为 fraction 的时候显示,输入框,0-1 的数字(两位小数),默认填充 0.2。 |
| 自定义基因集 | 当基因选择方法为 custom 的时候显示。 |
| 基因集打分方法 | Seurat|UCell。默认填充 UCell。 |
| reduction | pca|harmony,下拉框单选,默认 pca。 |
| K 分群数量 | 输入框,25-75 的整数,默认填充 25。 |
| Downsample | 默认填充 False,下拉框单选 True or False。 |
| Downsample_num | 默认填充 1000,数字,手动编辑填写(True 时显示)。 |
| 备注 | 自定义备注信息。 |
重要注意事项
CAUTION
- 大数据集处理:当细胞总数超过数万时,若
Downsample参数设置为False,分析可能会因内存不足而失败。强烈建议开启Downsample进行分析。 - Metadata 规范:请确保 RDS 文件中的 metadata 列名和内容不包含中文或特殊字符(如
&),否则可能导致流程错误。 - 物种匹配:确保选择的物种与实际数据匹配,否则会影响富集分析数据库的准确性。
操作流程
- 进入分析模块:在云平台导航至"高级分析"模块,选择"hdWGCNA"。
- 创建新任务:为您的分析任务命名,并选择要分析的样本或项目。
- 配置参数:根据上述指南,选择要分析的细胞类型、分组信息等。
- 提交任务:确认参数无误后,点击"提交"按钮,等待分析完成。
- 下载与查看:分析结束后,在任务列表中下载并查看生成的分析报告和结果文件。
结果解读
hdWGCNA 的分析报告包含丰富的图表和数据文件,以下是对核心结果的详细解读。
结果文件列表
| 文件名 | 内容说明 |
|---|---|
*_KME_modules.csv | 模块中基因的 KME(模块成员度)值,反映基因与模块的相关性。 |
*_cluster_allDMEs.csv | 不同细胞类型间模块特征基因的差异分析结果。 |
*_cluster_findallmarkers.csv | FindAllMarkers 识别的细胞类型标记基因。 |
*_hdWGCNA_findallmarkers_overlap_result.csv | hdWGCNA 模块基因与 FindAllMarkers 标记基因的重叠结果。 |
*_modules_GOenrich_result.txt | 各模块基因的 GO 功能富集分析结果。 |
modules_count.txt | 识别到的模块数量。 |
模块识别与特征分析
软阈值选择图
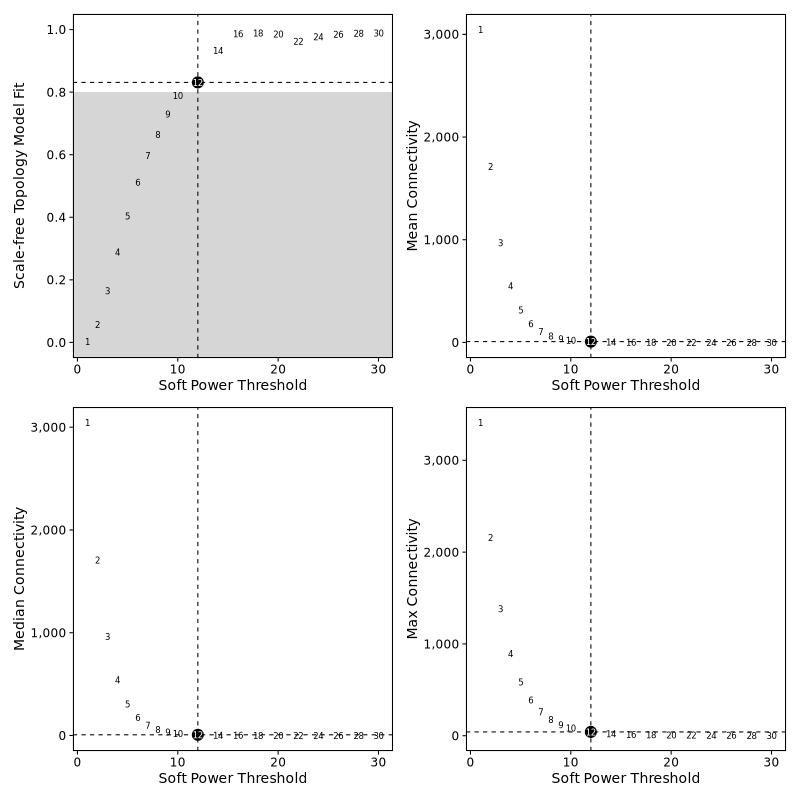
- 图表解读:展示不同软阈值下的网络连接性、平均连通性和拟合指数。
- 选择原则:选择拟合指数高且平均连通性适中的软阈值。
模块树状图
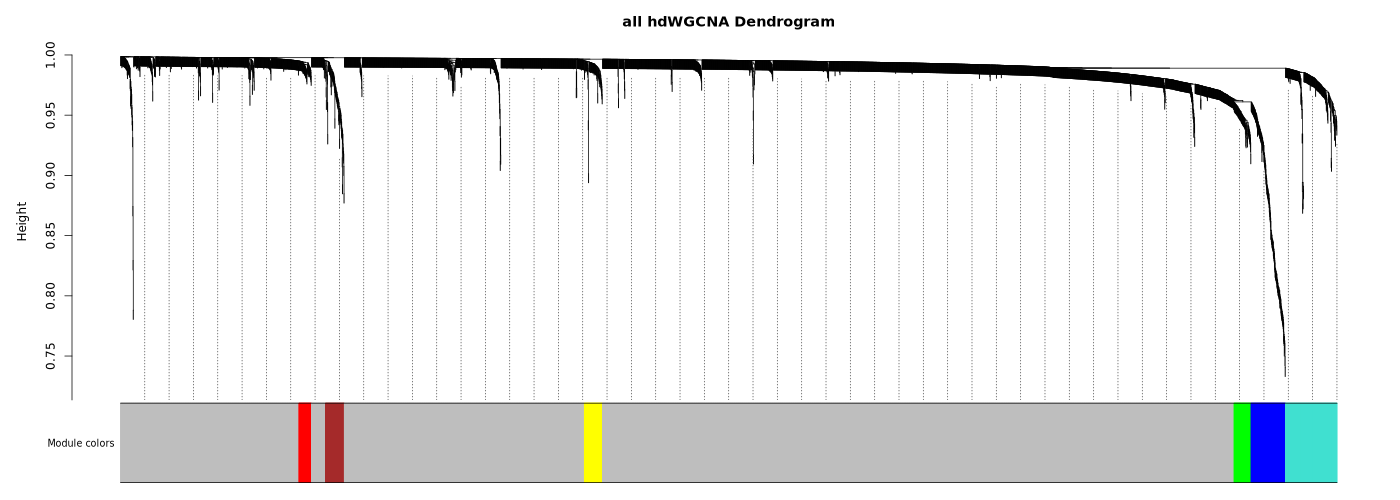
- 图表解读:展示基因层次聚类结果,不同颜色代表不同模块。
- 灰色模块:未被分配到任何模块的基因集合。
模块相关性热图
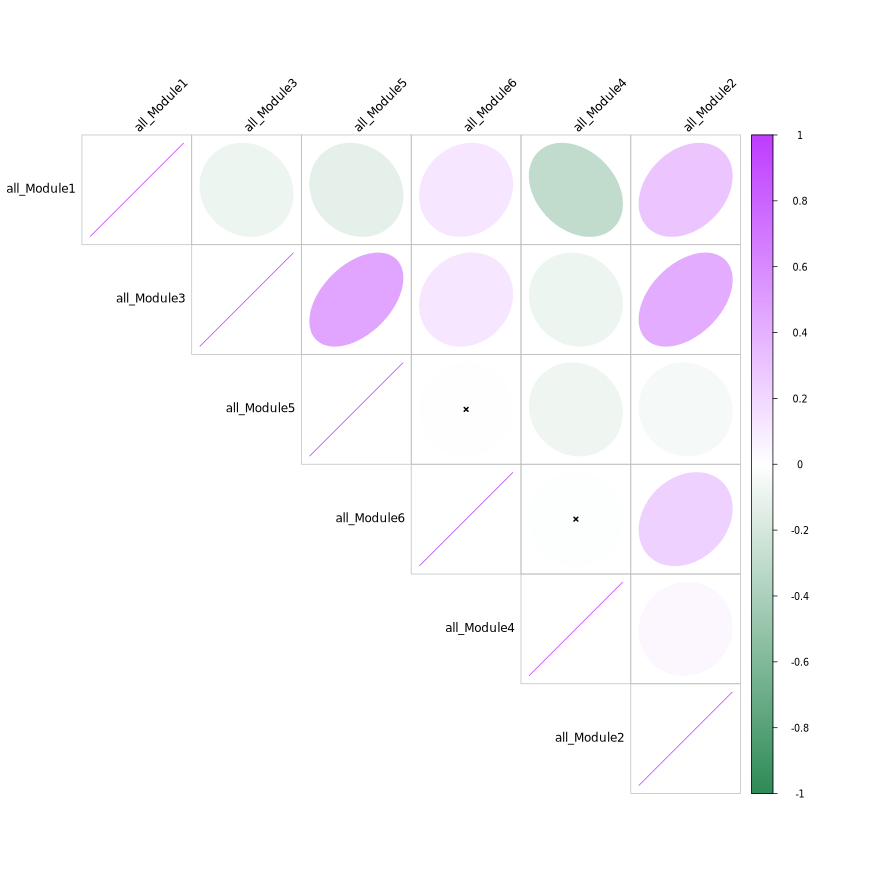
- 图表解读:展示不同模块特征基因间的相关性。
- 颜色含义:紫色表示正相关,绿色表示负相关。
模块功能分析
模块 KME 值图
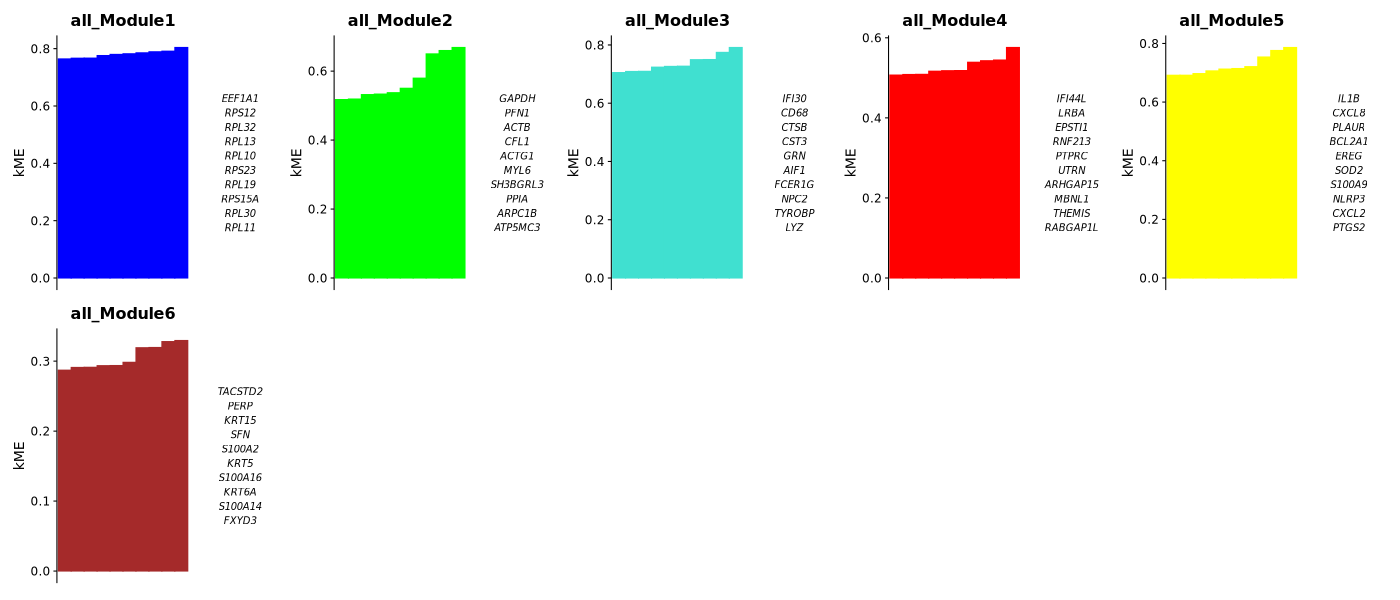
- 图表解读:展示每个模块中 KME 值最高的 10 个基因。
- KME 值含义:基因与模块特征基因的相关性,值越高表示基因越属于该模块。
模块网络图
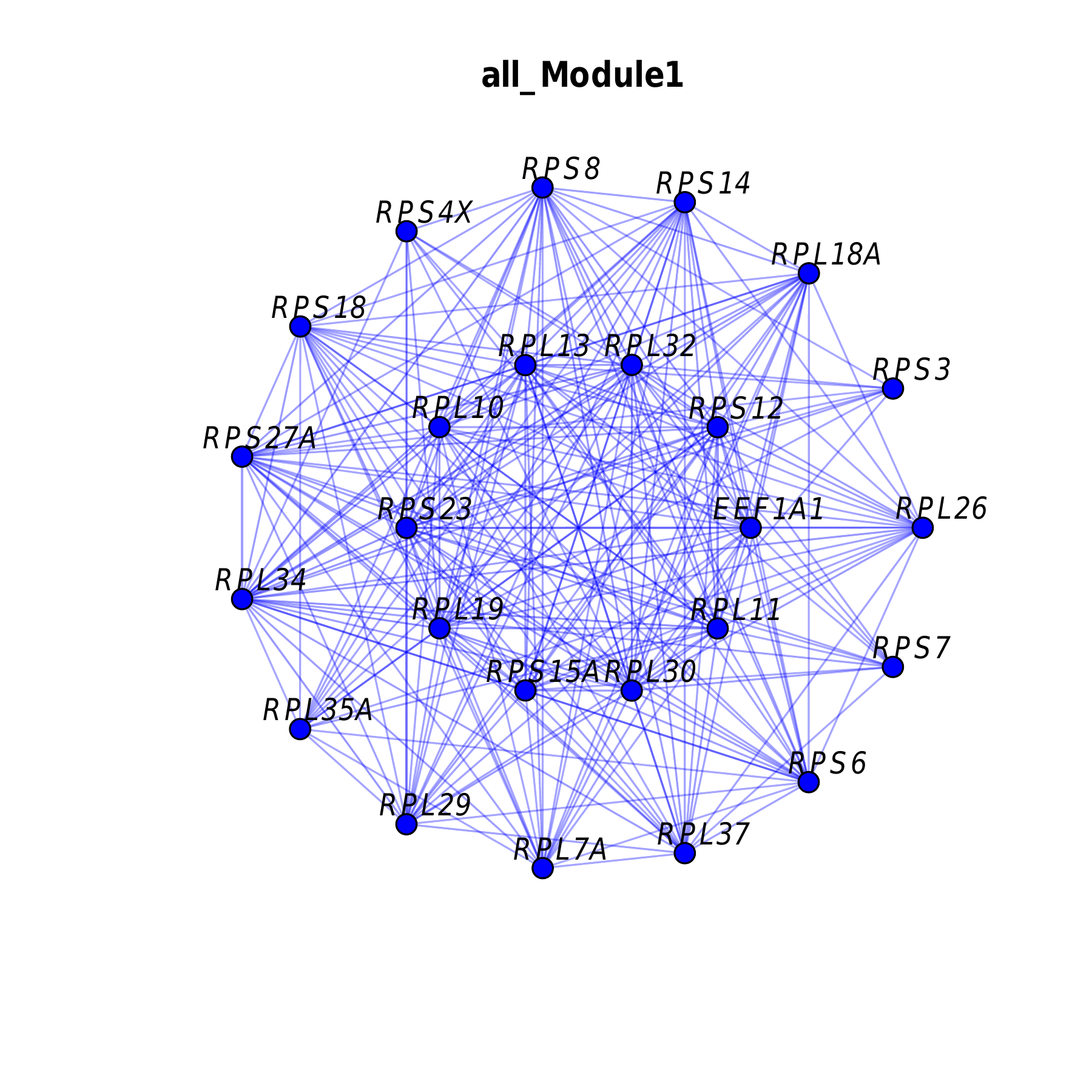
- 图表解读:展示模块中基因的共表达网络结构。
- 节点大小:表示基因的 KME 值。
- 连线粗细:表示基因间的连接强度。
模块活性可视化
UMAP 图
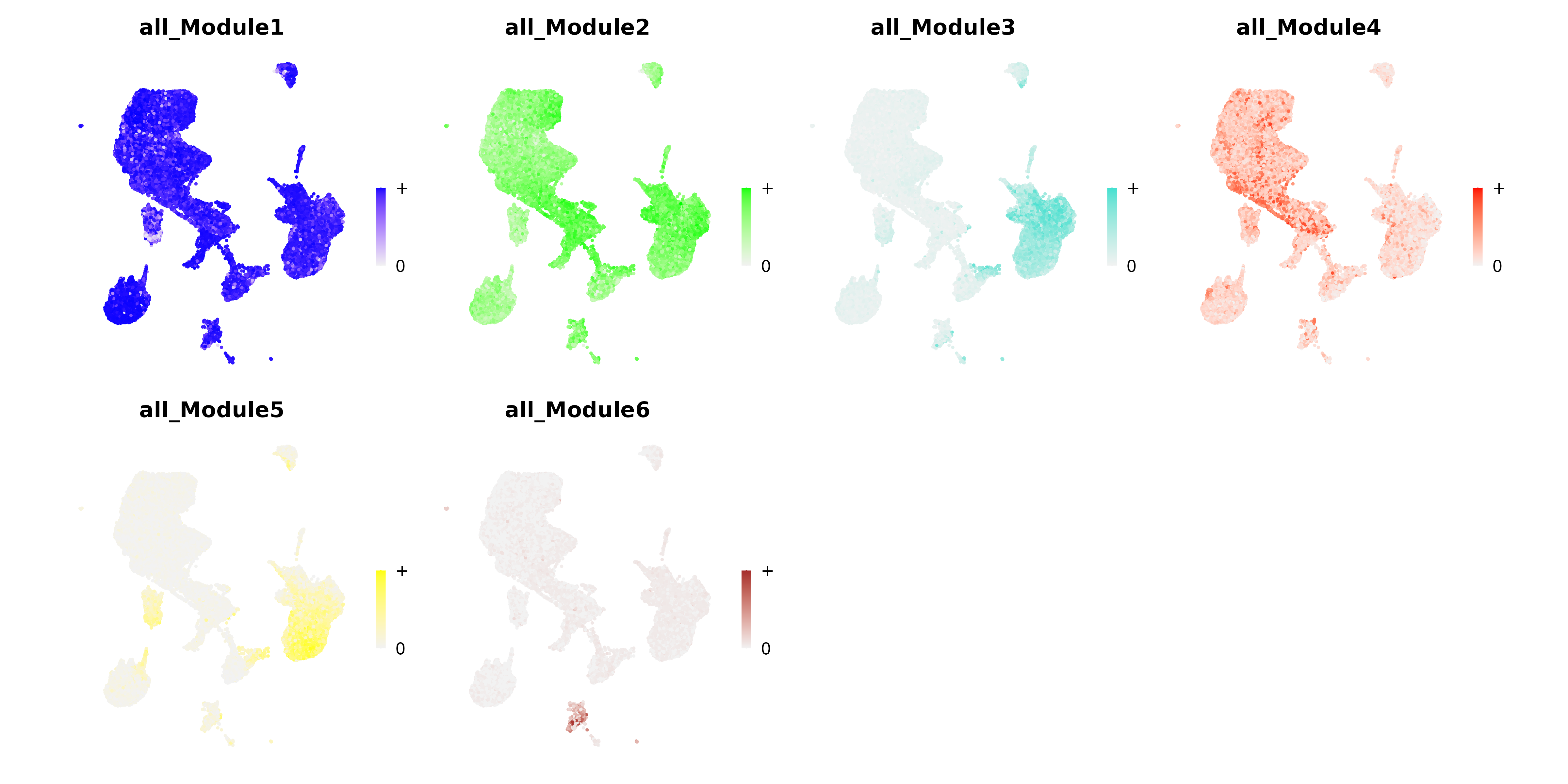
- 图表解读:在 UMAP 图上展示模块活性的空间分布。
- 颜色含义:深色表示高活性,浅色表示低活性。
小提琴图
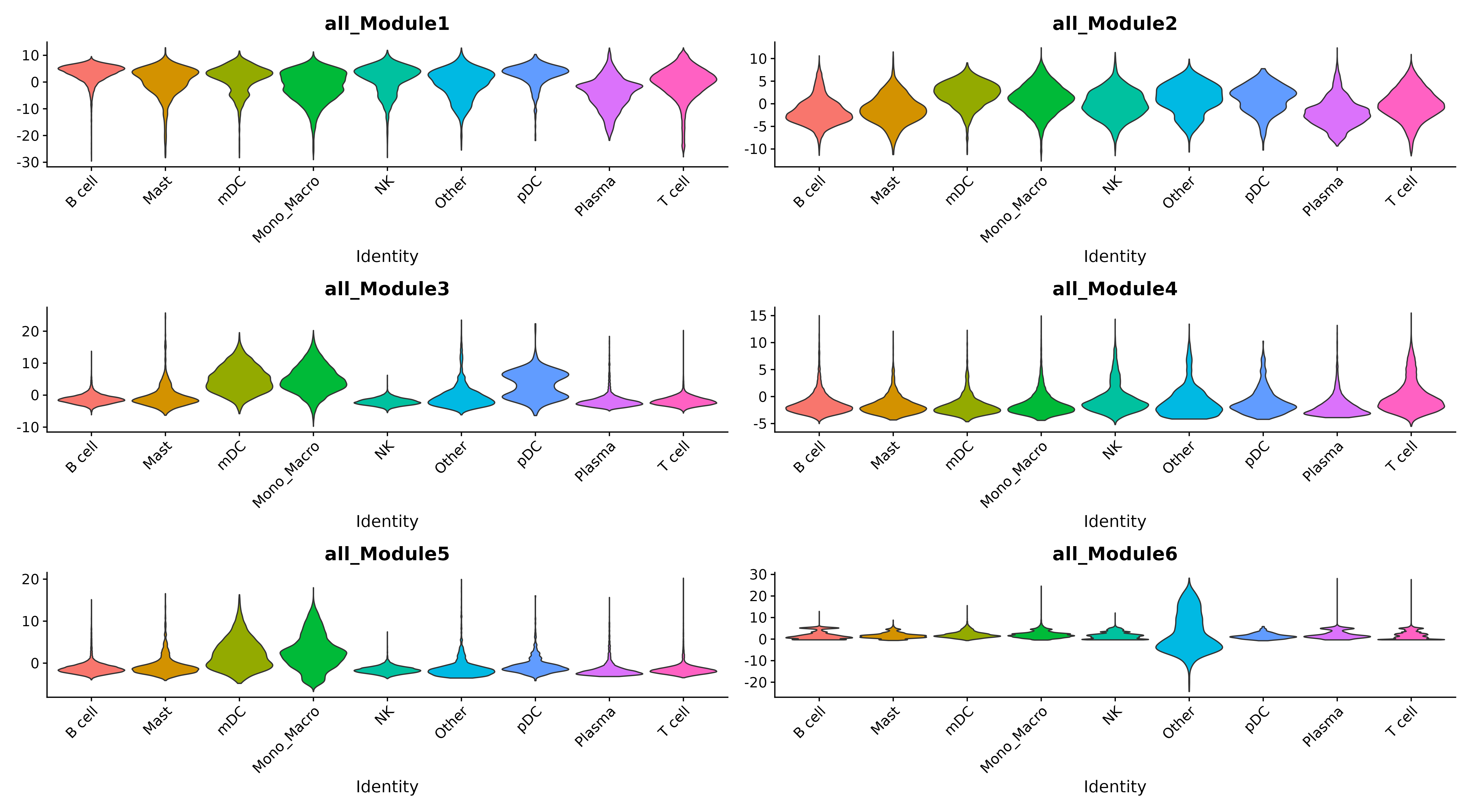
- 图表解读:展示不同细胞类型中模块活性的分布。
- 应用:识别细胞类型特异性的功能模块。
点图
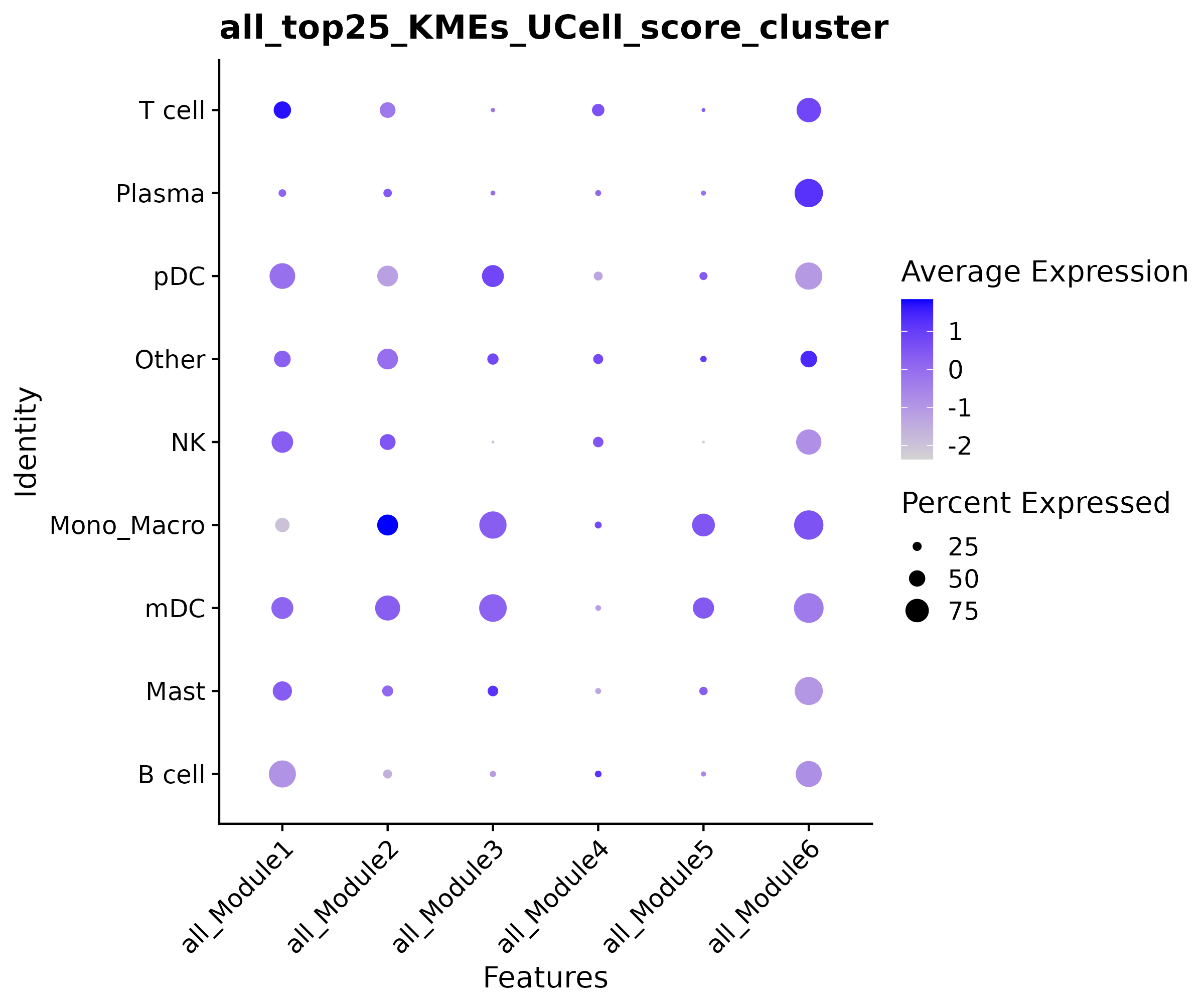
- 图表解读:展示不同细胞类型中模块活性的平均水平和表达比例。
- 颜色含义:蓝色表示高活性,浅色表示低活性。
- 点大小:表示基因在细胞类型中的表达比例。
模块差异分析
差异热图
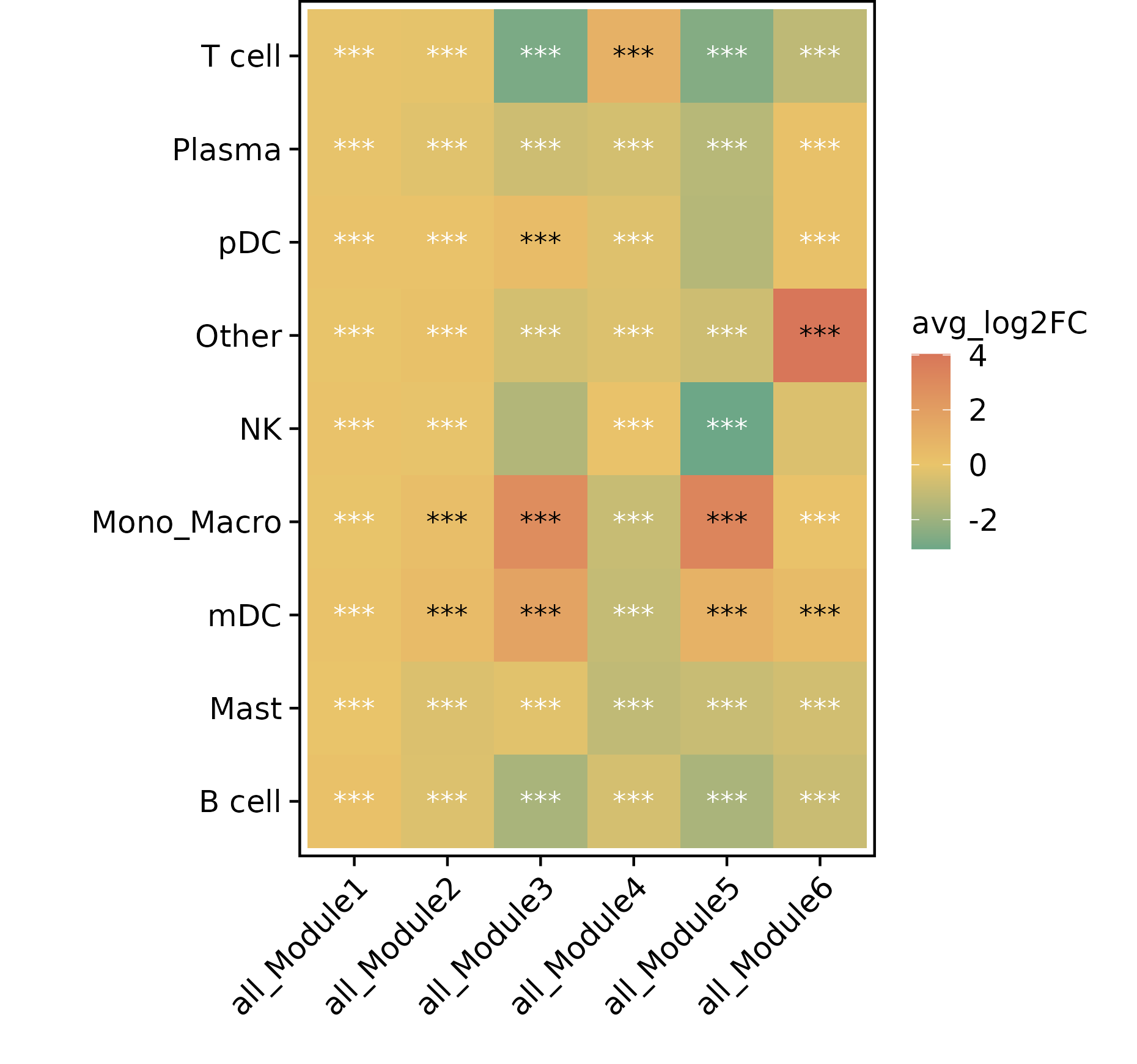
- 图表解读:展示不同细胞类型间模块活性的差异。
- 颜色含义:红色表示上调,绿色表示下调。
- 星号标注:表示差异显著性水平。
火山图
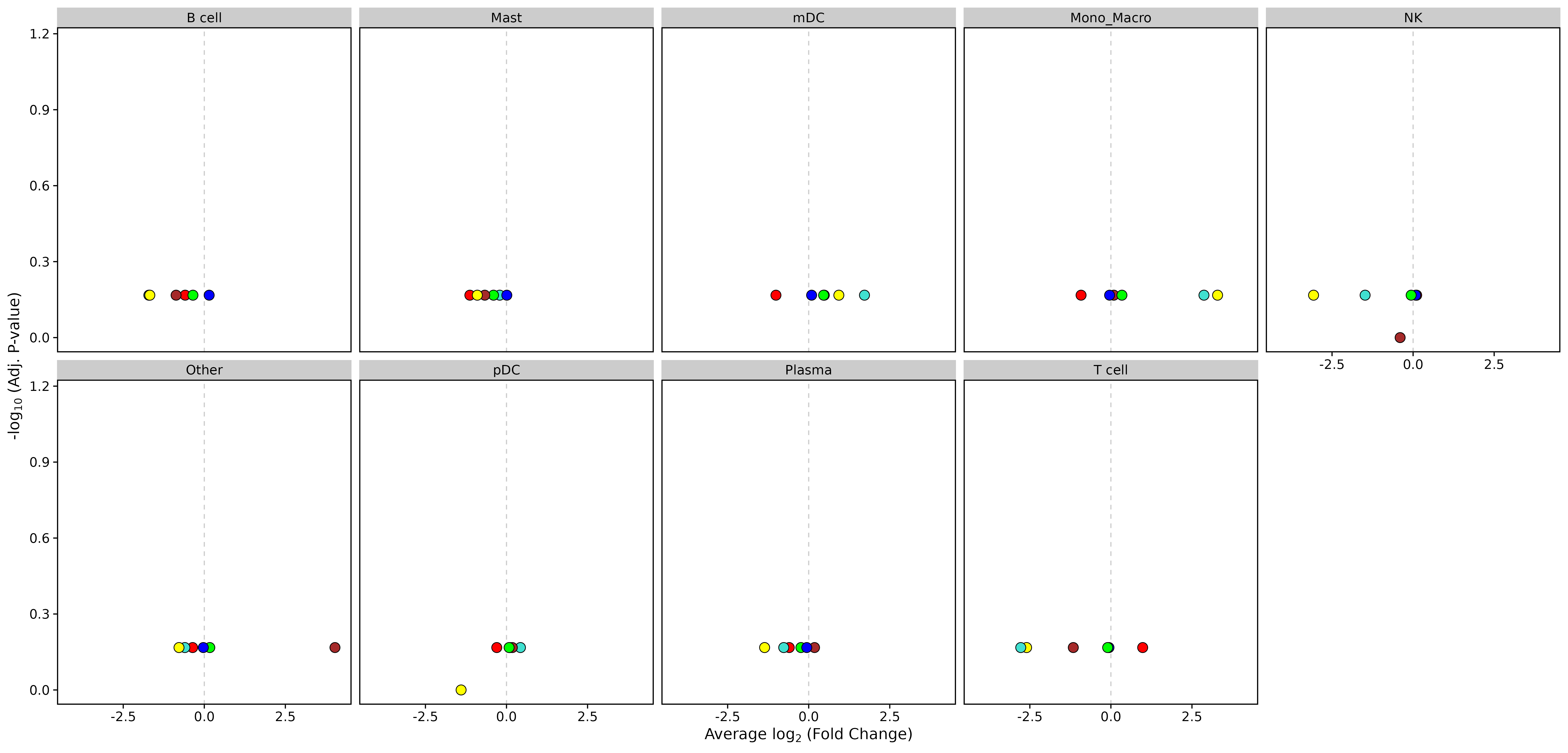
- 图表解读:展示不同细胞类型中模块活性的差异分析结果。
- 横轴:log2 倍数变化
- 纵轴:-log10(调整 p 值)
功能富集分析
GO 富集点图
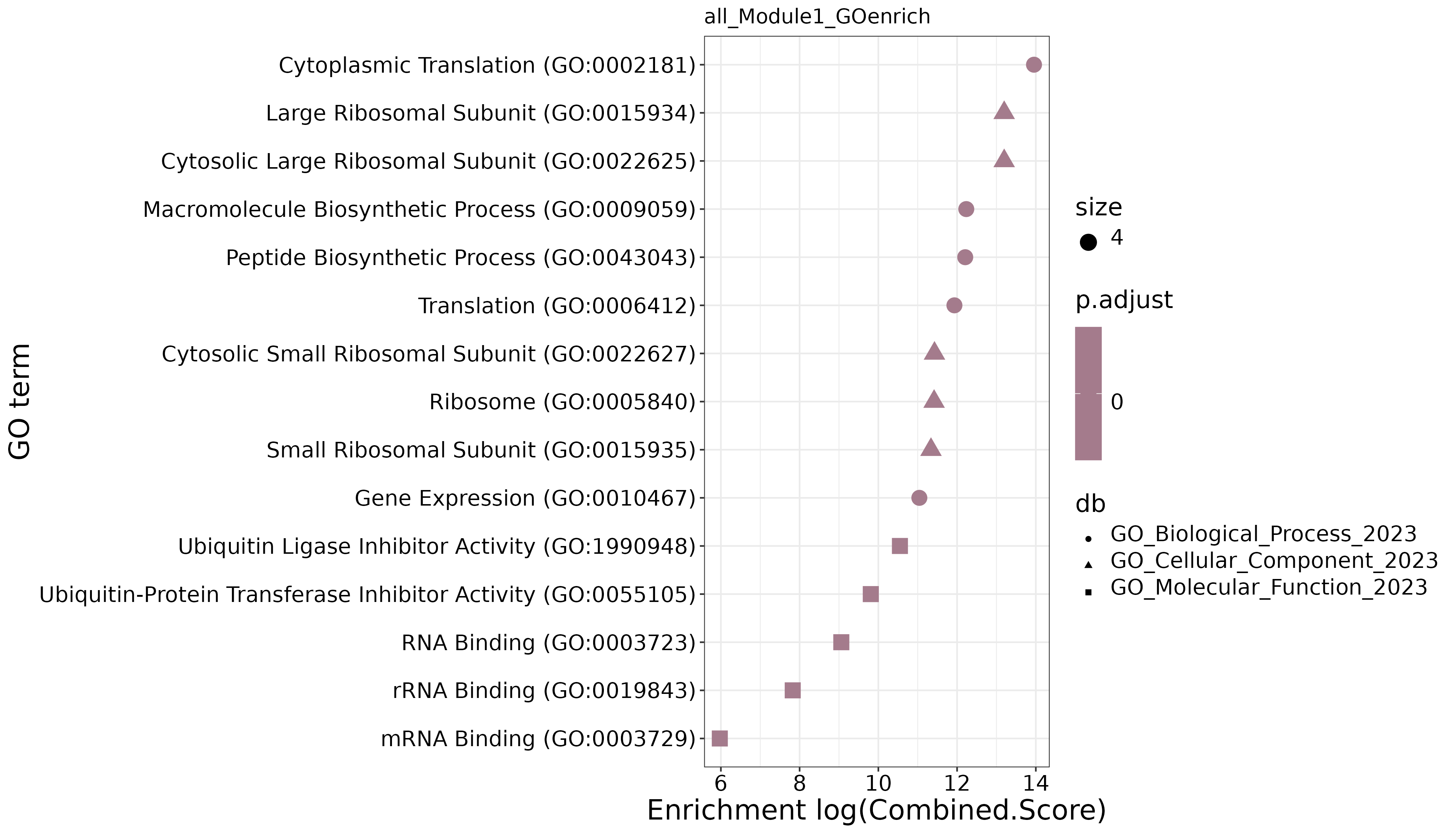
- 图表解读:展示模块基因的 GO 功能富集结果。
- 横轴:富集得分
- 纵轴:GO 条目
- 颜色:表示富集显著性
- 点大小:表示富集到该 GO 条目的基因数量
应用案例
案例一:在肿瘤微环境中识别免疫相关功能模块
- 文献:Morabito S, Miyamoto A, Pochareddy S, et al. bioRxiv. 2022.
- 背景:研究者希望识别肿瘤组织中与免疫逃逸及抑制相关的基因模块,重点关注肿瘤浸润 T 细胞和巨噬细胞亚群。
- 分析策略:对已注释的 T 细胞亚群和巨噬细胞亚群分别运行 hdWGCNA,选择
variable基因集并使用 UCell 进行模块活性打分;随后对模块进行 KME 筛选并做 GO/KEGG 富集分析,最后将模块活性在肿瘤与对照样本间进行差异分析。 - 核心发现:
- 识别到一个在肿瘤浸润巨噬细胞中特异性上调的“免疫抑制模块”,模块内包含 PD-L1(CD274)、IDO1 和若干代谢相关基因。
- 该模块在晚期样本中显著上调(差异分析 FDR < 0.05),并与肿瘤相关巨噬细胞标记基因高度重叠(KME>0.7)。
- GO/KEGG 结果提示该模块富集于“免疫抑制”、“色氨酸代谢”等通路,提示潜在的免疫调控机制可作为治疗靶点。
案例二:发育过程中特异性功能模块的鉴定
- 文献:Langfelder P, Horvath S. BMC Bioinformatics. 2008.
- 背景:研究胚胎发育过程中不同时间点间细胞类型的功能重编程,希望识别发育早期特异性模块。
- 分析策略:将细胞按时间点分组,使用
fraction方法选择在至少 20% 细胞中表达的基因,构建加权共表达网络并使用动态树切割识别模块;对模块进行时间序列的 Module Eigengene 分析以及富集分析。 - 核心发现:
- 发现一个在发育早期高表达的模块,含有多种干性维持相关基因(如 SOX2、NANOG 相关通路基因),其 Module Eigengene 随时间下降。
- 该模块的 GO 富集显示“干细胞维持”“细胞周期调控”相关条目,支持其在早期发育中的功能作用。
- 通过与单细胞注释结果重叠,证明该模块在祖细胞/干细胞亚群中富集。
案例三:药物处理响应相关模块的识别与验证
- 文献:Ricchiuti V, Li J, Shi J, et al. J Transl Med. 2021.
- 背景:比较处理组与对照组细胞以寻找药物响应相关的基因模块,并结合体外实验验证关键基因。
- 分析策略:合并处理组与对照组的 Seurat 对象,运行 hdWGCNA 并计算每个细胞的 Module Score;对模块活性在处理组与对照组间进行差异分析,同时筛选出高 KME 值基因用于后续实验验证。
- 核心发现与验证:
- 识别到一个显著上调的“应激响应模块”,模块内含若干药物代谢酶和应激相关转录因子。
- 选取模块中 KME 最高的 3 个基因进行 qPCR 和 Western blot 验证,实验结果与模块活性变化一致,证明了 hdWGCNA 结果的生物学可靠性。
注意事项与最佳实践
TIP
避免过度解读:hdWGCNA 结果是基于转录组数据的计算推断,不等于真实的调控关系。任何关键发现都需要后续的生物学实验来证实。
常见问题解答 (FAQ)
Q1: hdWGCNA 分析需要多长时间?
A: 分析时间取决于数据规模和计算资源配置。一般来说:
- 小数据集 (1,000-5,000 细胞):1-2 小时
- 中等数据集 (5,000-20,000 细胞):2-6 小时
- 大数据集 (>20,000 细胞):6-24 小时或更长 建议开启 Downsample 以加快分析速度。
Q2: KME 值和 Module Scores 的意义是什么?
A:
- KME (Module Membership):模块成员度,表示基因与模块特征基因的相关性。KME 值越高表示基因越属于该模块。
- Module Scores:模块得分,表示模块在单个细胞中的活性水平。通过 UCell 或 Seurat 方法计算得出。
Q3: 如何判断模块的生物学意义?
A: 可通过以下方法判断模块的生物学意义:
- 功能富集分析:通过 GO/KEGG 等富集分析了解模块的潜在功能
- 标记基因重叠:与已知的细胞类型标记基因进行重叠分析
- 差异分析:分析模块在不同细胞类型间的活性差异
Q4: 如何验证 hdWGCNA 分析结果的可靠性?
A: 可通过以下方式验证结果可靠性:
- 生物学验证:结合已知文献和数据库验证关键模块功能
- 实验验证:通过报告基因实验等方法验证关键模块基因
- 交叉验证:使用不同的数据集或分析方法验证结果一致性
参考资料
[1] LANGFELDER P, HORVATH S. WGCNA: an R package for weighted correlation network analysis[J]. BMC Bioinformatics, 2008, 9: 559.
[2] ZHANG B, HORVATH S. A general framework for weighted gene co-expression network analysis[J]. Stat Appl Genet Mol Biol, 2005, 4: Article17.
[3] MORABITO S, MIYAMOTO A, POCHAREDDY S, et al. Single-cell co-expression analysis identifies distinct regulatory programs in the developing human cortex[EB/OL]. bioRxiv, 2022.
[4] MILLER J A, CAI C, LANGFELDER P, et al. Strategies for aggregating gene expression data: the collapseRows R function[J]. BMC Bioinformatics, 2011, 12: 302.
[5] RICCHIUTI V, LI J, SHI J, et al. A practical guide to single-cell RNA sequencing for biomedical research and clinical applications[J]. J Transl Med, 2020.ransl Med. 2021 Sep 14;19(1):394. doi: 10.1186/s12967-021-02974-0. PMID: 34521462; PMCID: PMC8438992.